Water-oxidation catalysts mediate the four-electron, four-proton oxidation of water to dioxygen. In nature, the oxygen-evolving complex (OEC), a Mn4Ca cluster in the enzyme Photosystem II catalyzes this transformation. The energy of the resulting electrons and protons released during water oxidation is then stored as the biological equivalent of hydrogen. These reducing equivalents are eventually utilized for carbon dioxide fixation. The development of active and robust water-oxidation catalysts is crucial for the creation of artificial photosynthetic devices. The water splitting reaction is often considered to be one of the most challenging aspects of such a device, as the concerted removal of four protons and four electrons from two substrate waters is both energetically and kinetically demanding.
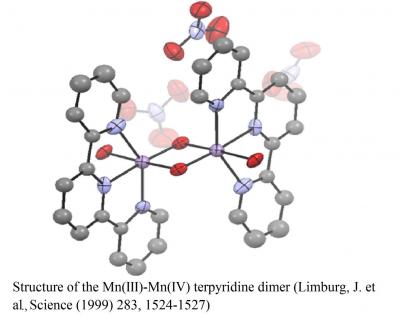
In collaboration with the Crabtree Group, we have developed several catalysts for water oxidation. Our work has led to the development of the first functional manganese model complex for the OEC of Photosystem II. When driven with primary oxidants such as hypochlorite or peroxymonosulfate (Oxone), our catalyst, the so-called “Mn-terpy dimer” effectively generates oxygen from water. Kinetic, spectroscopic, and electrochemical studies have provided rich details about the mechanism of oxygen evolution. Although there are many possible mechanisms for homogeneous water oxidation, we often envision a key step in which a high-valent metal-oxo species undergoes nucleophilic attack by substrate water:
[M=O]n+ + H2O –> M(n-2)+ + O2 + 2H+ + 2e-
Current research includes the development of new manganese complexes to act as structural and functional models of the OEC, as well as the synthesis and characterization of new heterogeneous manganese materials for the same reaction.
Other work in the group focuses on Cp* iridium complexes, which can be activated to form water-oxidation catalysts upon the addition of the chemical oxidant cerium(IV), sodium periodate, or an electrode potential. Depending on the ligand environment of the Cp*Ir precursor, the active catalyst generated can be either homogeneous or heterogeneous. Once oxidized, they become highly active catalysts capable of high rates of oxygen evolution with low overpotentials. On-going experiments are aimed at elucidating mechanistic details for iridium-catalyzed oxygen evolution, as well as incorporating these catalysts into more elaborate artificial photosynthetic systems.
References
“Functional Models for the Oxygen-Evolving Complex of Photosystem II”, Clyde W.Cady, Robert H. Crabtree and Gary W. Brudvig (2008) Coord. Chem. Rev. 252, 444-455.
“Water Oxidation Chemistry of Photosystem II”, Gary W. Brudvig (2008) Phil.Trans. R. Soc. Lond. B 363, 1211-1219.
“A Functional Model for O-O Bond Formation by the O2-Evolving Complex inPhotosystem II”, Julian Limburg, John S. Vrettos, Louise M. Liable-Sands, Arnold L. Rheingold, Robert H. Crabtree and Gary W. Brudvig (1999) Science 283,1524-1527.
“Highly Active and Robust Cp* Iridium Complexes for Catalytic Water Oxidation”, Jonathan F. Hull, David Balcells, James D. Blakemore, Christopher D. Incarvito, Odile Eisenstein, Gary W. Brudvig and Robert H. Crabtree (2009) J. Am. Chem.Soc. 131, 8730–8731.
“Anodic Deposition of a Robust Iridium Water-Oxidation Catalyst from Organometallic Precursors”, James D. Blakemore, Nathan D. Schley, Gerard W. Olack, Christopher D. Incarvito, Gary W. Brudvig and Robert H. Crabtree (2011) Chemical Science 2, 1, 94-98.